Back to Publications
Andrew D. Powell and Richard Dawes, Calculating potential energy curves with fixed-node diffusion Monte Carlo: CO and N2, J. Chem. Phys. 145, 224308 (2016).
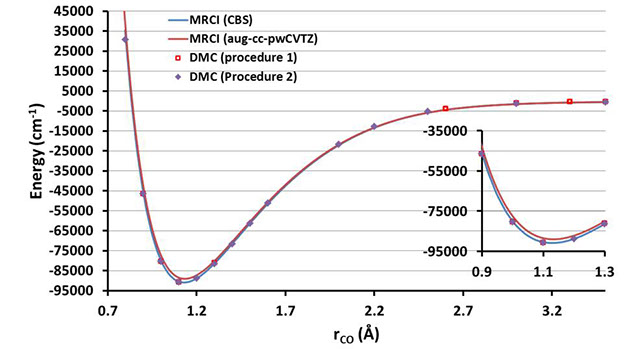
This study reports on the prospect for routine use of Quantum Monte Carlo (QMC) for the electronic structure problem, applying fixed-node Diffusion Monte Carlo (DMC) to generate highly-accurate Born-Oppenheimer potential energy curves (PECs) for small molecular systems. The singlet ground electronic states of CO and N2 were used as test cases. The PECs obtained by DMC employing multiconfigurational trial wavefunctions were compared with those obtained by conventional high-accuracy electronic structure methods such as multireference configuration interaction (MRCI) and/or the best available empirical spectroscopic curves. The goal was to test whether a straightforward procedure using available QMC codes could be applied robustly and reliably. Results obtained with DMC codes were found to be in close agreement with the benchmark PECs and the n3 scaling with the number of electrons (compared with n7 or worse for conventional high-accuracy quantum chemistry) could be advantageous depending on the system size. Due to a large pre-factor in the scaling, for the small systems tested here it is currently still much more computationally intensive to compute PECs with QMC. Nevertheless, QMC algorithms are particularly well-suited to large-scale parallelization and are therefore likely to become more relevant for future massively-parallel hardware architectures.