PES Fitting Methods
The philosophy behind the interpolative fitting methods that we use is ideally to render the fitting error negligible so that the computed dynamics or spectra directly represent the underlying electronic structure method.
The PES then acts only as an interface to make “direct dynamics” affordable. High-fidelity is particularly useful for spectroscopy applications where one might want to assess the individual contributions of small ab initio corrections to the spectra.
In a benchmark study of vibrational states of singlet methylene1 we fit a series of PESs each representing different basis sets and various single and multireference methods. Thus different complete basis set (CBS) extrapolation schemes could be tested using the separate PESs as components. Corrections such as core-correlation or the multireference Davidson correction were fit as separate surfaces which could be optionally added to determine their importance. In that same study a relatively low level ab initio method was used to compute a direct ab initio DVR grid of more than 22,000 points used to compute more than 200 bound states. Then results from three PESs fit to descending error tolerances were compared thus validating the idea of an interpolative PES as an efficient bridge to direct dynamics.
More recently a series of eight PESs for the CO-dimer system were computed in order to assess the importance of basis set completeness and correlation treatment by comparing CBS extrapolation schemes and the addition of core-electron correlation.2
Very recently four 9D PESs for methane were constructed incorporating nearly 100,000 ab initio data at the (AE)-MRCI-F12(Q)/CVQZ-F12 level.3 Three non-interpolative fits (PIP, NN, PIP-NN) all produced accurate fits with small RMS fitting errors (3-5 cm-1). However, the discrepancies between levels obtained by full-9D variational calculations and those measured experimentally were even smaller for the low lying levels, making it difficult to evaluate the electronic structure method due to convolution of fitting error. An interpolative fit with negligible error was used to benchmark the electronic structure method.
A description of our automated fitting methods can be found in our paper on the nitrous oxide dimer system.4 A review article has been submitted to Molecular Physics and should appear in early 2016.
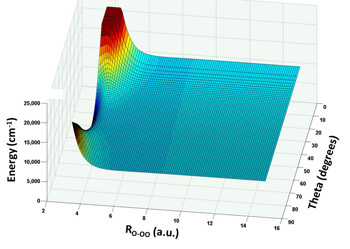
Plot of new PES for ozone showing the electronic energy for approach of an O-atom to an O2 molecule as a function of distance and angle.5,6 The minimum energy path for approach is monotonically attractive, but is repulsive for most angles creating a dynamical bottleneck.
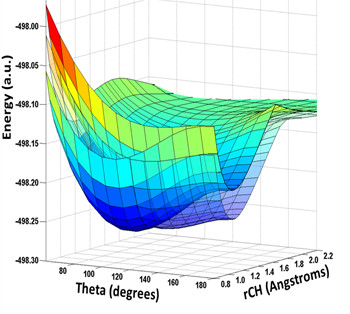
Plot of three PESs for CHCl, showing how the triplet state (transparent) passes through the ground singlet state, which also meets the excited singlet, becoming degenerate for linear geometries. The spectroscopy is perturbed by spin-orbit and Renner-Teller couplings.
Papers
1 Richard Dawes, Albert F. Wagner, and Donald L. Thompson. "Ab Initio Wavenumber Accurate Spectroscopy: 1CH2 and HCN Vibrational Levels on Automatically Generated IMLS Potential Energy Surfaces." J. Phys. Chem. A 113, 4709 (2009).
2 Richard Dawes, Xiao-Gang Wang and Tucker Carrington Jr., The CO dimer: a new potential energy surface and rovibrational calculations, J. Phys. Chem. A, 117, 7612 (2013).
3 Moumita Majumder, Samuel E. Hegger, Richard Dawes, Sergei Manzhos, Xiao-Gang Wang, Tucker Carrington Jr., Jun Li, and Hua Guo, Explicitly-correlated MRCI-F12 potential energy surfaces for methane fit with several permutation invariant schemes and full-dimensional vibrational calculations, Molecular Physics, 113, 1823, (2015).
4 Richard Dawes, Xiao-Gang Wang, Ahren W. Jasper, and Tucker Carrington Jr. "Nitrous oxide dimer: A new potential energy surface and rovibrational spectrum of the nonpolar isomer." J. Chem. Phys. 133, 134304 (2010).
5 Richard Dawes, Phalgun Lolur, Jianyi Ma and Hua Guo, Highly Accurate Ozone Formation Potential and Implications for Kinetics, J. Chem. Phys. 135, 081102 (2011).
6 Richard Dawes, Phalgun Lolur, Anyang Li, Bin Jiang and Hua Guo, An accurate global potential energy surface for the ground state of ozone, J. Chem. Phys. 139, 201103 (2013).